Research
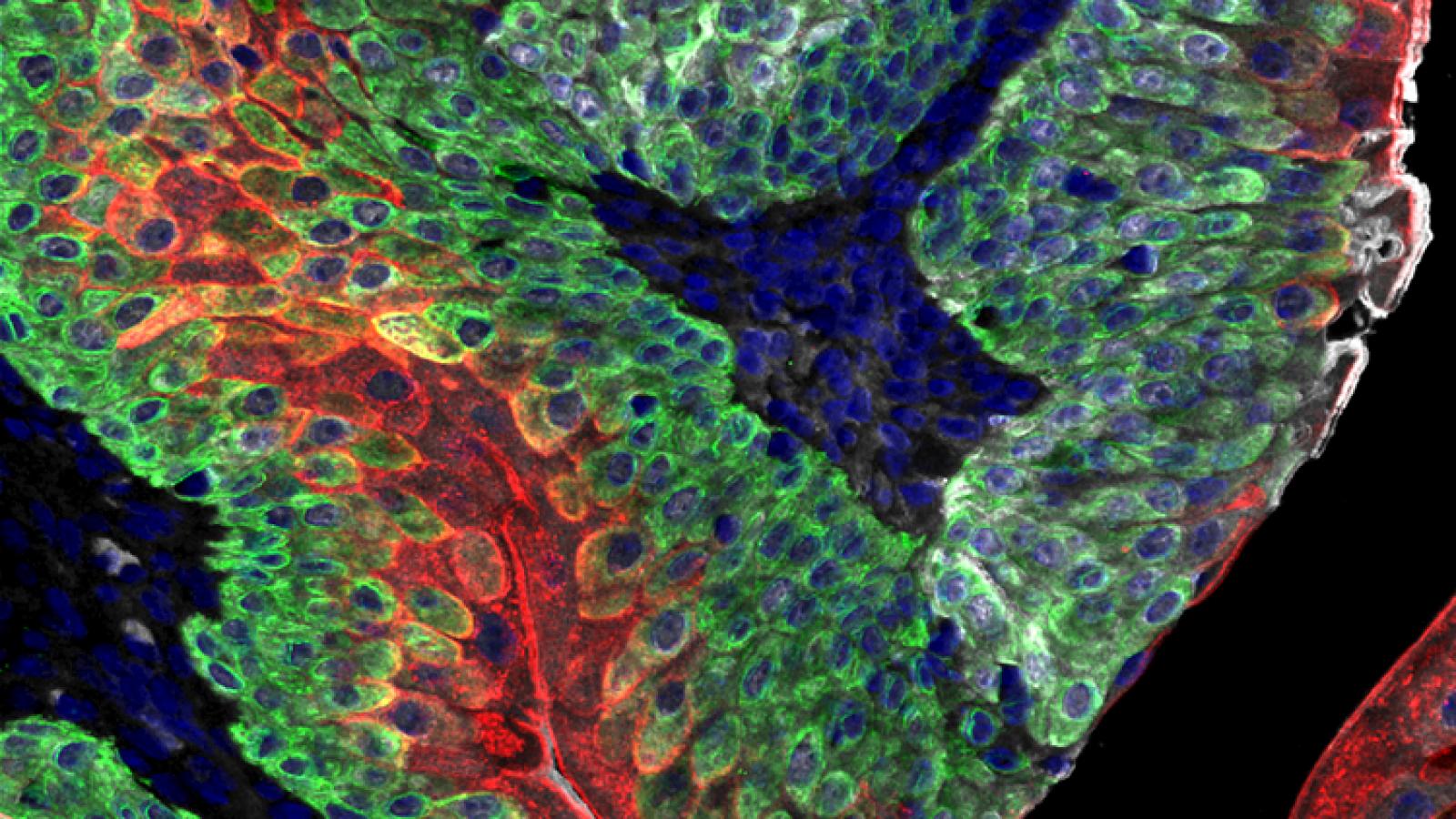
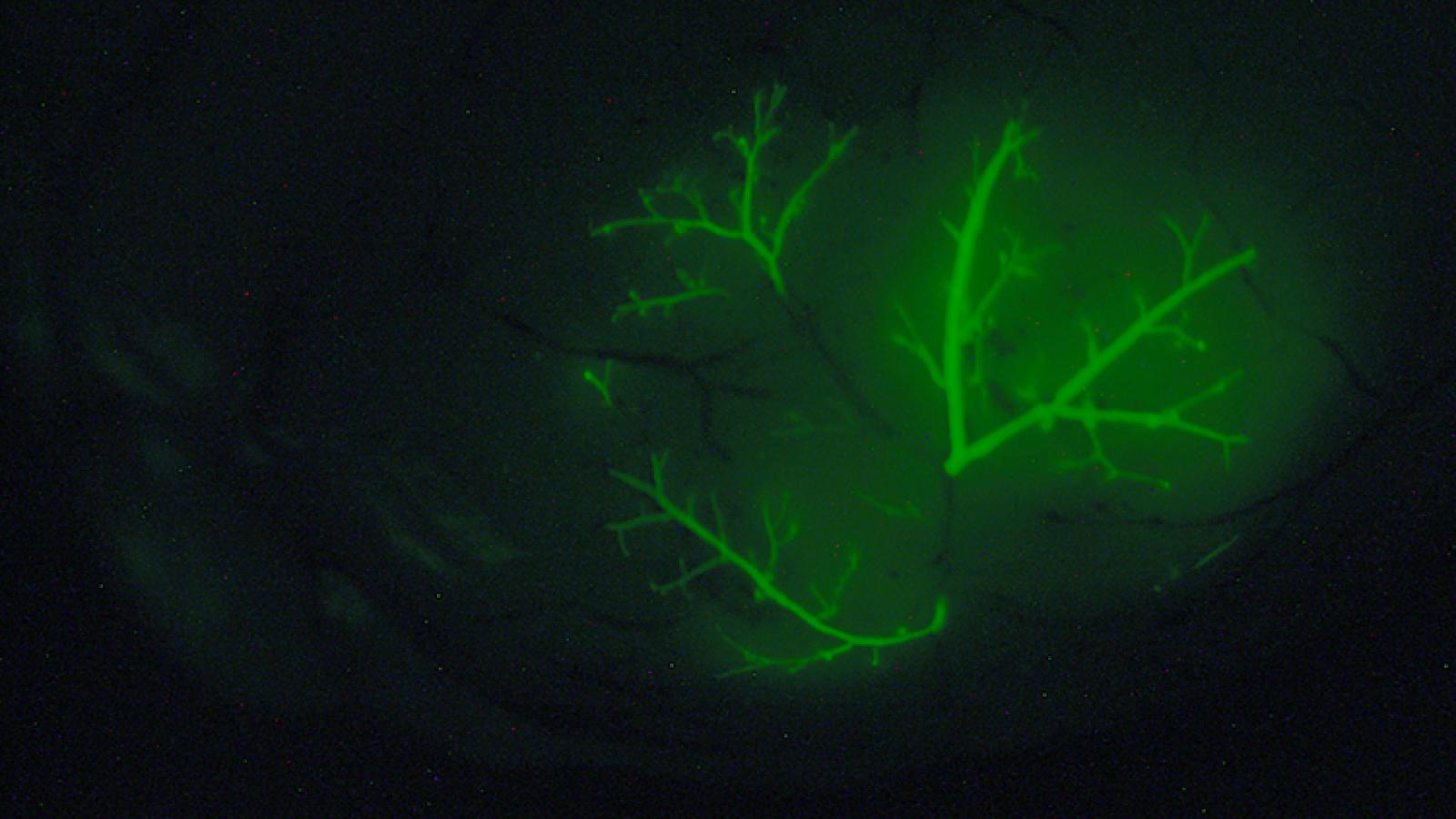


Our laboratory has a longstanding interest in understanding the relationship between the processes that control normal development and those that go awry in cancer (Abate-Shen, 2002; Le Magnen et al, 2018). Over the years, our laboratory has taken a multidisciplinary approach to investigating mechanisms of cancer initiation and progression using in vivo mouse models. We have generated unique genetically engineered mouse models (GEMMs) that have provided novel insights into mechanisms of tumorigenesis and valuable preclinical models for analysis of new cancer treatments and prevention.
Our major research focus has been on prostate and bladder cancer, which are highly complementary genitourinary malignancies. (See our reviews on prostate and bladder cancer.) In particular, the prostate and bladder have similar embryological origins from the primitive urogenital sinus and both have a high propensity to develop cancer particularly as a consequence of aging or environmental stresses such as inflammation. The vast majority of prostate and bladder cancers are relatively indolent, and unlikely to result in lethality; however, prostate and bladder cancers that progress to advanced disease often result in fatality. Moreover, most prostate and bladder cancer deaths occur from metastasis, which have eluded effective therapies. It is also now appreciated that many advanced cancers evolve into alternative cancer subtypes, such as neuroendocrine types, which arise from lineage plasticity. Thus, our research aims to elucidate cancer “drivers” or “master regulators” that control specific cancer phenotypes, including those that distinguish pre-malignant versus malignant disease, those that underlie drug response and drug resistance, those that promote metastasis, or those that define aberrant cancer subtypes. Overall, we seek to understand how normal mechanisms of transcriptional regulation and cellular differentiation can be coopted to give rise to distinct cancer subtypes. Toward this end, our work encompasses mechanism-based studies, analyses of genetically-engineered mouse models (GEMMs), and state-of-the-art systems biology approaches to understand these biological mechanisms.
Investigations of Prostate Cancer Specification and Cancer Initiation
Nowadays, many newly diagnosed cases of prostate cancer are relatively indolent and unlikely to result in death. Some, however, will eventually grow and spread through the body with deadly consequences. Yet it is difficult to distinguish the relatively few aggressive prostate tumors from the many indolent ones at early diseases stages when they might be mostly effectively treated. We have been using genetically engineered mouse models (GEMMs) to decipher mechanisms of cancer initiation and to identify biomarkers that distinguish aggressive cancer subtypes at early disease stages.
More than two decades ago, we, in collaboration with Michael Shen, identified the NKX3.1 homeobox gene as the earliest known marker of prostate epithelial differentiation. We showed that loss-of-function of Nkx3.1 in mutant mice leads to defective prostate development and predisposes to prostate cancer (Bhatia-Guar et al., 1999). Over the years, our analyses of NKX3.1 has provided key insights regarding the relationship between prostatic epithelial differentiation and cancer initiation. In particular, we have shown that NKX3.1 is necessary for appropriate prostate epithelial differentiation, while findings led by Michael Shen’s group have shown that Nkx3.1 marks a population of luminal prostate stem cells that can serve as a cell of origin for prostate cancer (Wang et al., 2009).
One of our most exciting findings is that expression NKX3.1 is sufficient to re-specify a non-prostatic epithelium to fully differentiate into prostate in vivo, still one of the few such examples in which a single gene can convert a differentiated epithelium to an alternative cell fate (Dutta et al., 2016). Loss of function of NKX3.1 also promotes cellular plasticity following inflammation as occurs during prostatitis (Le Magnen et al, 2018). This is associated with promotion of cancer initiation and leads to increased susceptibility to oxidative damage, whereas gain of function of NKX3.1 protects against oxidative stress (Ouyang et al., 2005). Recently, we have discovered a novel role for NKX3.1 as a transcription factor in mitochondria in response to oxidative stress, where it directly regulates mitochondrially-encoded genes. We showed that low expression levels of NKX3.1 combined with low expression of mitochondrial ETC genes are associated with adverse clinical outcome, whereas high levels of mitochondrial NKX3.1 protein are associated with favorable outcome. Our findings uncover a non-nuclear function for NKX3.1 that is a key mechanism for suppression of prostate cancer. We are now investigating whether analyses of the expression levels and sub-cellular localization of NKX3.1 in patients at risk of cancer progression can improve risk assessment in a precision prevention paradigm, particularly for men undergoing active surveillance (Papachristodoulou et al., 2021).
We have also investigated Nkx3.1 mutant mice as a model of indolent prostate cancer, which led to the identification of a three-gene biomarker panel that distinguishes indolent from aggressive prostate cancer (Irshad et al., 2013). In related work, our co-clinical investigations correlating the status of NKX3.1 with response to a common chemoprevention agent, 5α-reductase inhibitors (5-ARIs) have shown that reduced NKX3.1 expression is predictive of response to 5-ARI (Dutta et al., 2018). All together, our investigations of the NKX3.1 homeobox gene have shed new light on mechanisms that promote prostatic integrity in normal circumstances and how they go awry in cancer initiation, and suggest new options to improve early intervention for prostate cancer patients.

Investigations of Advanced Prostate Cancer
While most men diagnosed with early stage prostate cancers are now essentially curable, the prognosis for those with advanced disease is still not favorable. Advanced prostate cancer is often associated with a transition to castration-resistance, in some cases with neuroendocrine differentiation, which is highly aggressive, usually metastatic, and often fatal (Shen and Abate-Shen 2010). In collaboration with Michael Shen’s group, we have been addressing these clinical challenges through the generation of an extensive series of GEMMs based on inducible deletion of tumor suppressors or inducible activation of oncogenes specifically in the prostate epithelium (Wang et al, 2012; Floc'h et al, 2012; Aytes et al., 2013; Zou et al., 2017; Arriaga and Abate-Shen 2018; Arriaga et al., 2020; Giacobbe and Abate-Shen 2021). All of our GEMMs are freely available from the Jackson laboratory (Abate-Shen and Pandolfi 2013).
These GEMMs recapitulate the various stages of advanced prostate cancer, including castration resistance, neuroendocrine differentiation and lethal metastasis. Our GEMMs of advanced prostate cancer are based on loss-of-function of the Pten tumor suppressor gene. We have modeled Pten loss in combination with various other alleles including activation of RAF or RAS, which leads to metastatic prostate cancer (Wang et al, 2012; Aytes et al., 2013, 2017) or loss of function of p53, which leads to castration resistance that progresses to neuroendocrine prostate cancer (Zou et al., 2017). In particular, lineage-tracing of neuroendocrine prostate cancer in a GEMM based on loss of Pten and p53 provided direct genetic evidence that treatment-induced neuroendocrine prostate cancer arises via transdifferentaition, which is a specialized type of lineage plasticity (Zou et al, 2017).
Our GEMMs of metastatic prostate cancer display fully penetrant metastatic prostate cancer and shares molecular features in common with lethal prostate cancer in humans (Aytes et al., 2013). Lineage-tracing of metastases in this GEMM in vivo, combined with molecular investigations, led to the identification of master regulators of metastasis, including potential targets for therapeutic intervention (Aytes et al., 2018). Most notably, by refining our GEMM of metastatic prostate cancer we have now generated a model of highly penetrant bone metastasis. Molecular analyses of the bone metastases reveal that co-activation of MYC and RAS pathways promotes prostate cancer metastasis, and have led to the identification of a gene signature – called META-16 – that is prognostic for time to metastasis and predictive of treatment response in human patient cohorts. Thus, analysis of bone metastasis in this unique mouse model has uncovered conserved mechanisms with potential translational significance (Arriaga and Abate-Shen 2018).
Another major focus of our current research has been on applying systems biology approaches to integrate molecular insights from our GEMMs into the study of human cancer. In collaboration with the laboratories of Andrea Califano and Michael Shen, we have generated “interactomes” (or regulatory networks) for both mouse and human prostate cancer, using them to pursue cross-species analyses. These studies have led to the identification of synergistic regulators of advanced prostate cancer that are robust biomarkers of its most aggressive subtype (Aytes et al., 2014). We have also used this approach to identify drivers of drug response and drug resistance (Mitrofanova et al., 2015) (Zou et al, 2017), with the ultimate goal of “personalizing” more effective therapeutic management of drug response and resistance for patients.
In our most recent studies, we address the challenges of prioritizing treatments for individual cancer patients by introducing OncoLoop, a precision medicine framework that predicts drug sensitivity in human tumors and their pre-existing high-fidelity models by leveraging drug perturbation profiles. OncoLoop is a transcriptomic-based experimental and computational framework that can support rapid-turnaround co-clinical studies to identify and validate drugs for individual patients, which can then be readily adapted to clinical practice. This framework should be applicable in many cancer contexts for which appropriate models and drug perturbation data are available (Vasciaveo et al., 2023).
Modeling Invasive Bladder Cancer
Bladder cancer is the fifth most common cancer occurring worldwide and a major cause of morbidity and mortality. While superficial bladder cancer, which accounts for ~75% of patients, have good clinical outcome, the remaining ~25% of patients develop into invasive disease with high mortality. Although the landscape is changing with new options for immunotherapy and improved chemotherapy, there are still relatively curative treatments for metastatic bladder cancer, and currently available strategies have only modestly improved survival over the past 20 years. Despite its significant clinical relevance, the molecular biology of bladder cancer has been relatively understudied, which is due in part to challenges in creating in vivo models of human disease (Kobayashi et al., 2015).
Some time ago, we described a GEMM that progresses from non-muscle invasive to muscle invasive bladder cancer, which was based on surgical delivery of an Adenovirus expressing Cre recombinase into the bladder lumen of mice with conditional floxed alleles for the Pten and Trp53 (Puzio-Kuter et al., 2009). Co-clinical analyses of chemotherapy response in this GEMM provided the rationale for an informative clinical trial for patients with high-risk non-muscle invasive bladder cancer demonstrating the relevance of this GEMM as a preclinical model of bladder cancer (DeCastro et al., 2020).
We have since developed second-generation GEMMs of bladder cancer with improved specificity of Cre-mediated recombination in bladder urothelium. Using two independent approaches to target gene recombination to specific cell types in the bladder urothelium, we found that inactivation of Pten and p53 in basal, but not non-basal, cells is sufficient for progression from NMIBC to MIBC. Cross-species analysis comparing the mouse gene signature of early bladder cancer with a corresponding human bladder cancer signature identified a prognostic signature for MIBC, supporting the relevance of these GEMMs for studying human bladder cancer and introducing a robust gene signature that may help to stratify patients at risk for developing MIBC (Park et al., 2021).